Publications
 Open |
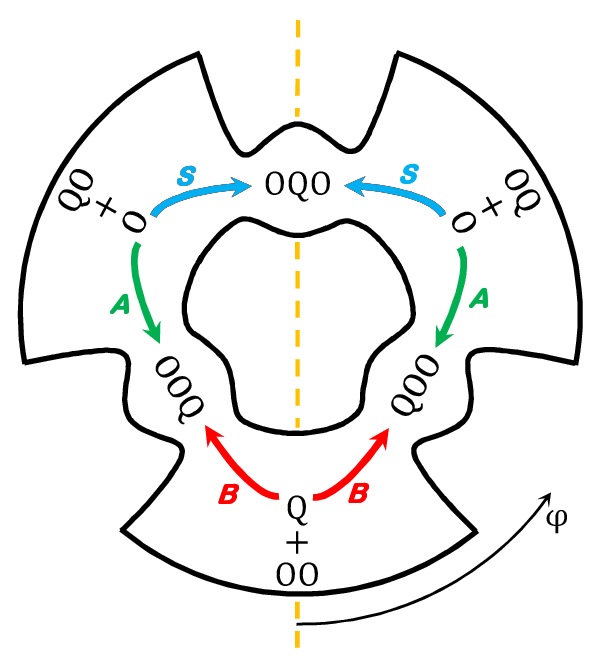
Several Levels of Theory for Description of Isotope Effects in Ozone: Symmetry Effect and Mass Effect
Teplukhin A. and Babikov D. J. Phys. Chem. A, DOI: 10.1021/acs.jpca.8b09025 (2018) The essential components of theory for the description of isotope effects in recombination reaction that forms ozone are presented, including the introduction of three reaction pathways for symmetric and asymmetric isotopomers, a brief review of relevant experimental data for singly- and doubly-substituted isotopologues, the definitions of ζ-effect and η-effect, and the introduction of isotopic enrichment δ. Two levels of theory are developed to elucidate the role of molecular symmetry, atomic masses, vibrational zero-point energies and rotational excitations in the recombination process. The issue of symmetry is not trivial, since the important factors, such as ½ and 2, appear in seven different places in the formalism. It is demonstrated that if all these effects are taken into account properly, then no anomalous isotope effects emerge. At the next level of theory, a model is considered in which one scattering resonance (sitting right at the top of centrifugal barrier) is introduced per ro-vibrational channel. It is found that this approach is equivalent to statistical treatment with partition functions at the transition state. Accurate calculations using hyper-spherical coordinates show that no isotope effects come from difference in the number of states. In contrast, differences in vibrational and rotational energies lead to significant isotope effects. However, those effects appear to be local, found for the rather extreme values of rotational quantum numbers. They largely cancel when rate coefficients are computed for the thermal distribution of rotational excitations. Although large isotope effects (observed in experiments) are not reproduced here, this level of theory can be used as a foundation for more detailed computational treatment, with accurate information about resonance energies and lifetimes computed and included. |
 Open |
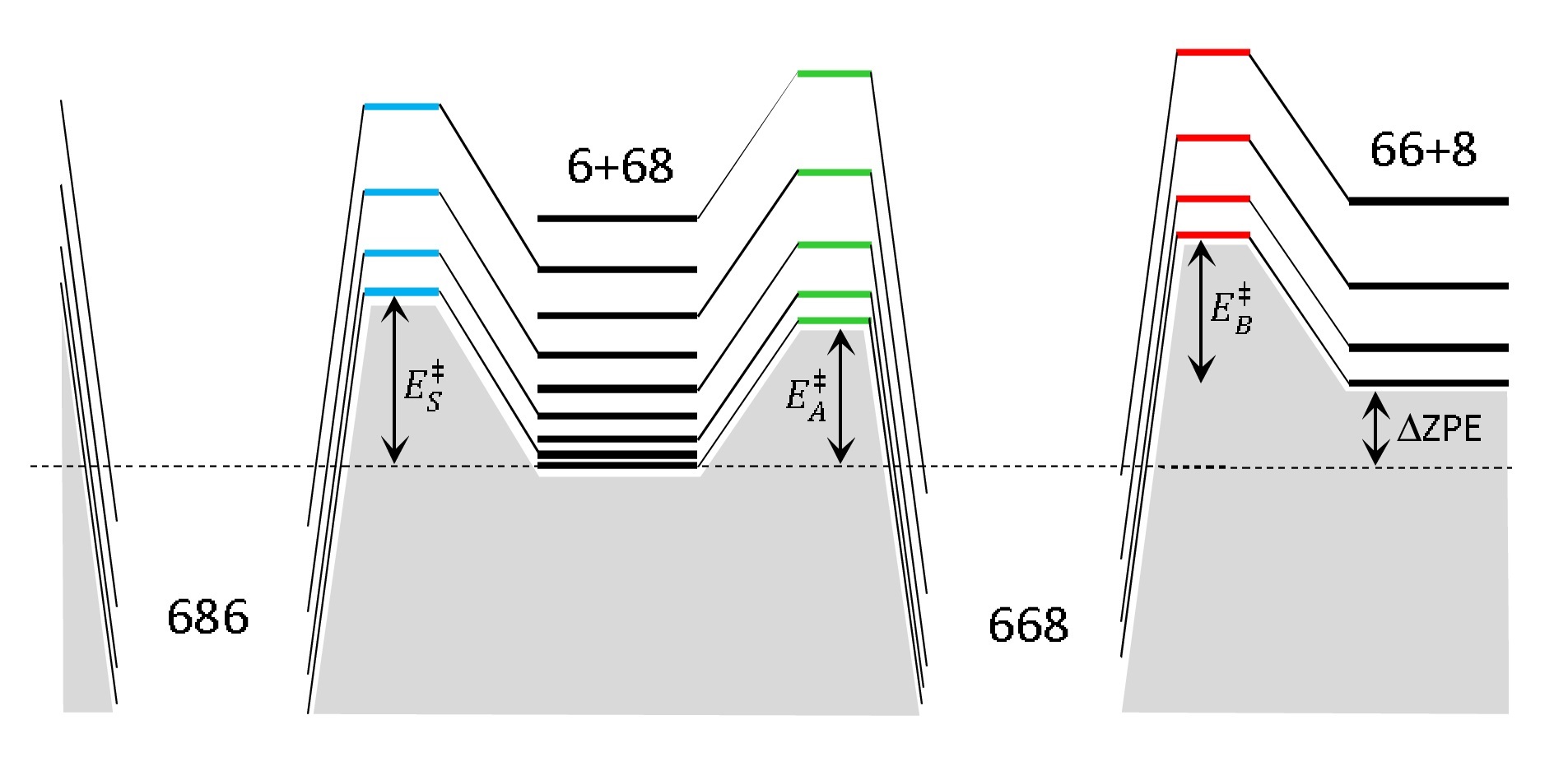
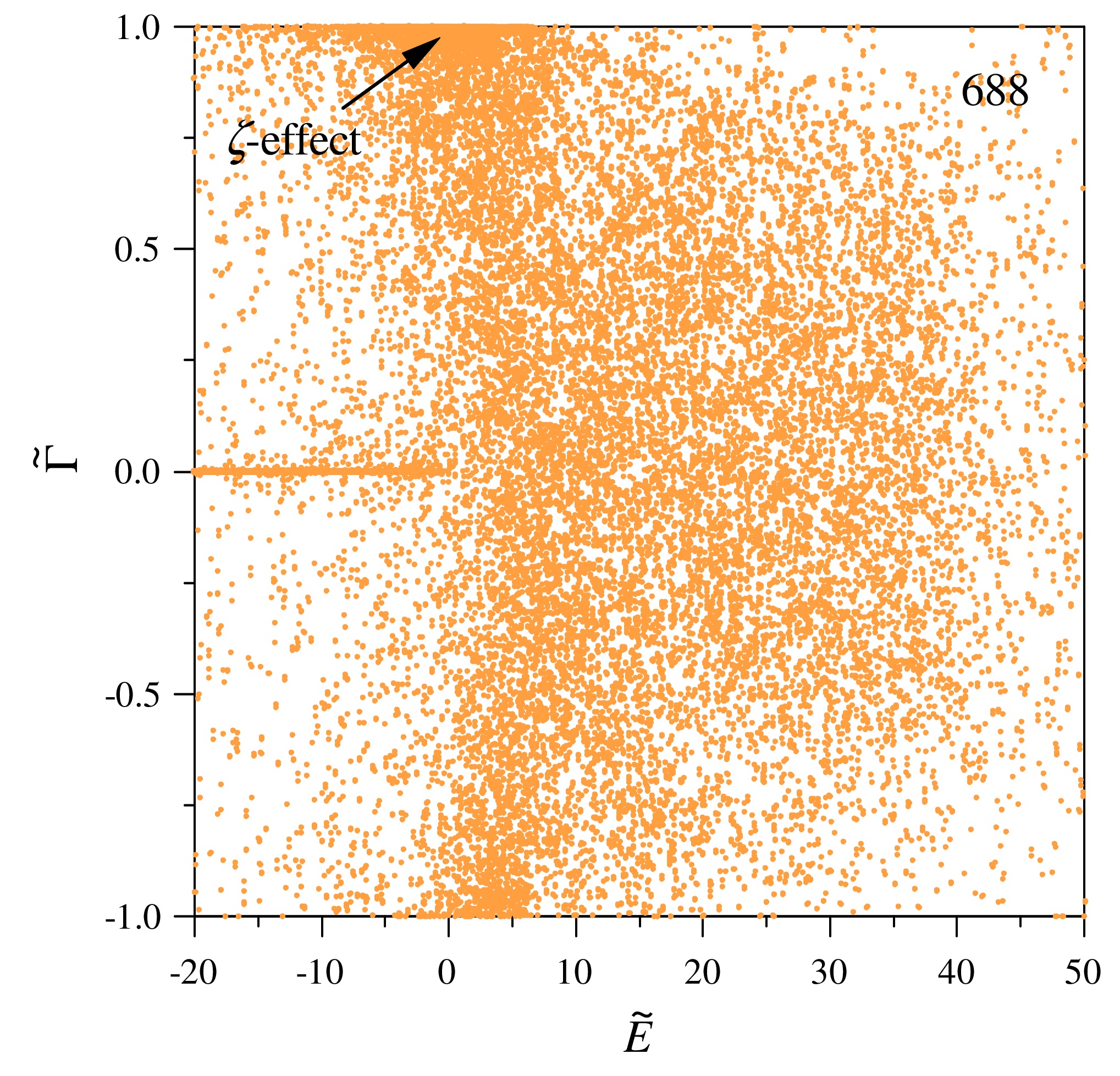
Several levels of theory for description of isotope effects in ozone: Effect of resonance lifetimes and channel couplings
Teplukhin A., Gayday I. and Babikov D. J. Chem. Phys., 149, 164302 (2018) In this paper, two levels of theory are developed to determine the role of scattering resonances in the process of ozone formation. At the lower theory level, we compute resonance lifetimes in the simplest possible way, by neglecting all couplings between the diabatic vibrational channels in the problem. This permits to determine the effect of “shape” resonances, trapped behind the centrifugal barrier and populated by quantum tunneling. At the next level of theory, we include couplings between the vibrational channels, which permits to determine the role of Feshbach resonances and interaction of different reaction pathways on the global PES of ozone. Pure shape resonances are found to contribute little to the overall recombination process since they occur rather infrequently in the spectrum, in the vicinity of the top of the centrifugal barrier only. Moreover, the associated isotope effects are found to disagree with experimental data. By contrast, Feshbach-type resonances are found to make dominant contribution to the process. They occur in a broader range of spectrum, and their density of states is much higher. The properties of Feshbach resonances are studied in detail. They explain the isotopic ζ-effect, giving theoretical prediction in good agreement with experiments for both singly and doubly substituted ozone molecules. Importantly, Feshbach resonances also contribute to the isotopic η-effect, moving theoretical predictions in the right direction. Some differences with experimental data remain, which indicates that there may be another additional source of the η-effect. |
 Open |
Properties of Feshbach and “shape”-resonances in ozone and their role in recombination reactions and anomalous isotope effects
Teplukhin A. and Babikov D. Faraday Discuss., DOI: 10.1039/c8fd00089a (2018) Computational modelling of recombination reactions that form ozone require the inclusion of several quantum mechanical effects such as symmetry, zero-point energy, scattering resonances and tunneling. Major elements of theory for rigorous description of this process are reviewed, with emphasis on interpreting the famous anomalous isotope effect due to substitutions of 18O. Three reaction pathways, for the formation of symmetric and asymmetric isotopologues of ozone, are introduced and a hierarchy of theory levels is outlined. Lower levels of theory are used to account for the effects of symmetry, isotope mass, rotational excitations and vibrational zero-point energy differences. They happen to be equivalent to statistical descriptions of the process and do not show anomalous isotope effects. Properties of scattering resonances should be included at the next level of theory, and may finally explain the isotope effect. Shape resonances, trapped behind the centrifugal barrier and populated by tunneling, can be studied by neglecting couplings between the diabatic ro-vibrational states of the system. Inclusion of these couplings enables the formation of Feshbach resonances. Accurate calculations using hyper-spherical coordinates are performed to obtain resonance energies, lifetimes and wavefunctions. Differences between the shape resonances and Feshbach resonances are emphasized. |
 Open |
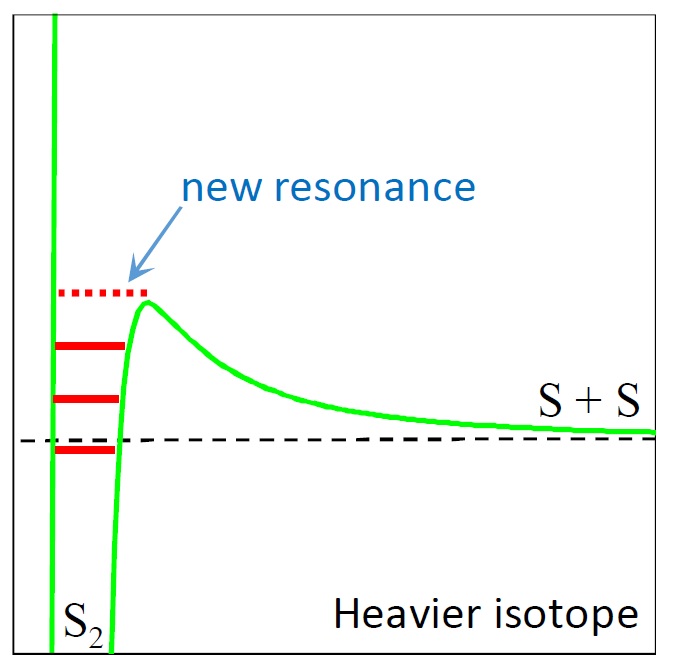
One possible source of mass-independent fractionation of sulfur isotopes in the Archean atmosphere of Earth
Babikov D., Semenov A. and Teplukhin A. Geochim. Cosmochim. Acta, 204, 388 (2017) Energy transfer mechanism for recombination of two sulfur atoms into a diatomic molecule, S2, is studied theoretically and computationally to determine whether the rate coefficient of this process can be significantly affected by isotopic substitutions, and whether the resultant isotope effect is expected to be mass-dependent or mass-independent. This is one of sulfur polymerization processes thought to be important in the anoxic atmosphere of the Archean Earth and, potentially, relevant to mass-independent fractionation of sulfur isotopes. A simplified theoretical approach is employed, in which all properties of S2 molecule are characterized rather accurately, whereas the process of stabilization of metastable S2* by bath gas collisions is described approximately. Properties of individual scattering resonances in S2 are studied in detail, and it is found that most important contributions to the recombination process come from ro-vibrational states formed near the top of centrifugal barrier, and that the number of such states is about 50 (in 32S32S). Absolute value of recombination rate coefficient is computed to be 1.22 × 10−33 cm6/s (for 32S32S at room temperature and atmospheric pressure), close to experimental result. Two distinct isotope effects are identified. One is a classical mass-dependent effect due to translational partition function, which leads to a weak, smooth, and negative mass-dependence of rate coefficient (4% decrease when the mass is raised from 32S32S to 34S34S). Second effect, due to quantized resonances, is two orders of magnitude stronger, but is local. In practice, due to presence of multiple individual resonances, this phenomenon leads to irregular mass-independent variations of rate coefficients in the ranges ±5%. It is also demonstrated that in real molecules this irregular behavior is expected to be somewhat smoother, and the isotope effect is somewhat smaller, due to dependence of stabilization cross section on properties of individual resonances (not described by present model). Thus, additional calculations of stabilization cross sections are needed in order to give quantitative prediction of this mass-independent isotope effect, and to determine its relevance to mass-independent fractionation of sulfur isotopes in the Archean rock record. |
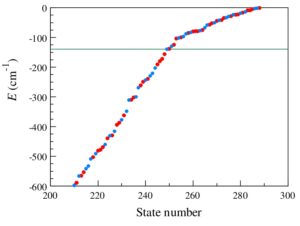 Open |
Efficient method for calculations of ro-vibrational states in triatomic molecules near dissociation threshold: Application to ozone
Teplukhin A. and Babikov D. J. Chem. Phys., 145, 114106 (2016) A method for calculations of rotational-vibrational states of triatomic molecules up to dissociation threshold (and scattering resonances above it) is devised, that combines hyper-spherical coordinates, sequential diagonalization-truncation procedure, optimized grid DVR, and complex absorbing potential. Efficiency and accuracy of the method and new code are tested by computing the spectrum of ozone up to dissociation threshold, using two different potential energy surfaces. In both cases good agreement with results of previous studies is obtained for the lower energy states localized in the deep (∼10 000 cm-1) covalent well. Upper part of the bound state spectrum, within 600 cm-1 below dissociation threshold, is also computed and is analyzed in detail. It is found that long progressions of symmetric-stretching and bending states (up to 8 and 11 quanta, respectively) survive up to dissociation threshold and even above it, whereas excitations of the asymmetric-stretching overtones couple to the local vibration modes, making assignments difficult. Within 140 cm-1 below dissociation threshold, large-amplitude vibrational states of a floppy complex O⋯O2 are formed over the shallow van der Waals plateau. These are assigned using two local modes: the rocking-motion and the dissociative-motion progressions, up to 6 quanta in each, both with frequency ∼20 cm-1. Many of these plateau states are mixed with states of the covalent well. Interestingly, excitation of the rocking-motion helps keeping these states localized within the plateau region, by raising the effective barrier. |
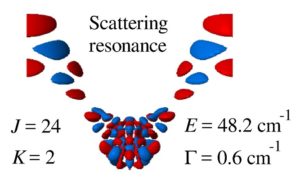 Download PDF |
A full-dimensional model of ozone forming reaction: the absolute value of the recombination rate coefficient, its pressure and temperature dependencies
Teplukhin A. and Babikov D. Phys. Chem. Chem. Phys., 18, 19194 (2016) Rigorous calculations of scattering resonances in ozone are carried out for a broad range of rotational excitations. The accurate potential energy surface of Dawes is adopted, and a new efficient method for calculations of ro-vibrational energies, wave functions and resonance lifetimes is employed (which uses hyper-spherical coordinates, the sequential diagonalization/truncation approach, grid optimization and complex absorbing potential). A detailed analysis is carried out to characterize distributions of resonance energies and lifetimes, their rotational/vibrational content and their positions with respect to the centrifugal barrier. Emphasis is on the contribution of these resonances to the recombination process that forms ozone. It is found that major contributions come from localized resonances at energies near the top of the barrier. Delocalized resonances at higher energies should also be taken into account, while very narrow resonances at low energies (trapped far behind the centrifugal barrier) should be treated as bound states. The absolute value of the recombination rate coefficient, its pressure and temperature dependencies are obtained using the energy-transfer model developed in the earlier work. Good agreement with experimental data is obtained if one follows the suggestion of Troe, who argued that the energy transfer mechanism of recombination is responsible only for 55% of the recombination rate (with the remaining 45% coming from the competing chaperon mechanism). |
 Download PDF |
Visualization of potential energy function using an isoenergy approach and 3D prototyping
Teplukhin A. and Babikov D. J. Chem. Educ., 92(2), 305 (2015) In our three-dimensional world, one can plot, see, and comprehend a function of two variables at most, V(x,y). One cannot plot a function of three or more variables. For this reason, visualization of the potential energy function in its full dimensionality is impossible even for the smallest polyatomic molecules, such as triatomics. This creates some barrier to understanding the interaction of atoms in a molecule. It would be beneficial to see all features of the global potential energy function at the same time (which can include deep covalent wells, transition states, shallow van der Waals wells, and reaction channels) without fixing or relaxing some degrees of freedom. In this paper, we review the isoenergy approach that allows visualization of the potential energy function of a triatomic molecule in its full dimensionality in 3D space as a volume, not as a surface. Also, we propose the use of 3D-printing capabilities to create plastic models of such isoenergy objects that can be taken into hands and inspected in detail from any perspective. |
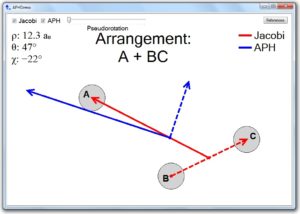 Download PDF |
Interactive tool for visualization of adiabatic adjustment in APH coordinates for computational studies of vibrational motion and chemical reactions
Teplukhin A. and Babikov D. Chem. Phys. Lett., 614, 99 (2014) The adiabatically-adjusting principal-axes hyperspherical (APH) coordinates reviewed in this letter are one of the best coordinate sets developed for computational treatment of spectroscopy and dynamics of triatomic molecules. Unfortunately, it is not so easy to understand and interpret them, compared to other simpler coordinates, like valence coordinates or Jacobi coordinates. To address this issue, we developed a desktop application called APHDemo. This tool visualizes the process of adjustment of the APH coordinates to the shape of a triatomic molecule during molecular vibrations or chemical reaction, and helps to understand their physical meaning without going into complicated math. |
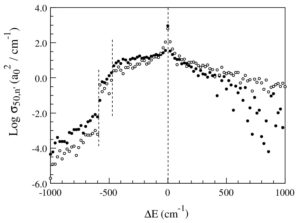 Download PDF |
Frozen rotor approximation in the mixed quantum/classical theory for collisional energy transfer: Application to ozone stabilization
Teplukhin A., Ivanov M. and Babikov D. J. Chem. Phys., 139, 124301 (2013) A frozen-rotor approximation is formulated for the mixed quantum∕classical theory of collisional energy transfer and ro-vibrational energy flow [M. Ivanov and D. Babikov, J. Chem. Phys. 134, 144107 (2011)]. Numerical tests are conducted to assess its efficiency and accuracy, compared to the original version of the method, where rotation of the molecule in space is treated explicitly and adiabatically. New approach is considerably faster and helps blocking the artificial ro-vibrational transitions at the pre- and post-collisional stages of the process. Although molecular orientation in space is fixed, the energy exchange between rotational, vibrational, and translational digresses of freedom still occurs, allowing to compute ro-vibrational excitation and quenching. Behavior of the energy transfer function through eight orders of magnitude range of values and in a broad range of ΔE is reproduced well. In the range of moderate -500 ≤ ΔE ≤ +500 cm-1 the approximate method is rather accurate. The absolute values of stabilization cross sections for scattering resonances trapped behind the centrifugal threshold are a factor 2-to-3 smaller (compared to the explicit-rotation approach). This performance is acceptable and similar to the well-known sudden-rotation approximation in the time-independent inelastic scattering methods. |